网络药理学:分子动力学模拟:gromacs和Charmm-GUI模拟(前置准备、正式模拟、RMSD/RMSF/回旋半径gyrate/氢键/自由能形貌图/轨迹动图/MMPBSA等计算、结果可视化)
25.03.19二次修订:因超算平台软件变更,module load anaconda/2022.10 修正为 module load miniforge/24.1.2
你需要知道的是:
- 薛定谔做分子对接,直接导出配体
mol2格式,蛋白pdb格式即可。不需要文本操作来分离蛋白和配体。 - 本人
MD全过程是使用北京超算平台gromacs做的,7w原子总长时间(前置准备+正式模拟+可视化分析)共3小时。 - 如果你的蛋白中含有锌离子或镁离子也是一样的过程,但如果你的蛋白口袋中含有铁离子等,在
3.2.1步骤改为选择相应索引即可。 - 本篇博客以
PDB ID为7OVV的蛋白,MOL004004配体来做演示。
常见问题:
为什么很多教程是使用文本操作来分离蛋白和配体,而不是软件直接导出呢?
这是因为用软件导出的话会在文件开头添加软件相关的注释和说明,但和蛋白信息其实无关。而这些信息可能导致手动搭建体系(也就是我薛定谔专栏第一篇博客讲的传统体系)出问题,所以必须用文本操作只保留关键的原子坐标信息。
那为什么需要先合并对接的构象和蛋白,然后再单独分离呢?
这是为了防止直接分离导致原子序数不一样。
一些命令行必备知识:
因为本文涉及到了超算平台,也就是linux平台上的操作,所以在此补充一下基本知识。
补全文件名/目录:在命令行窗口,按住tab键可以自动补全文件名或目录名。粘贴:在主机复制后,在超算平台等linux平台按住shift+ctrl+v进行粘贴,或者点击鼠标右键进行粘贴。复制:在超算平台等linux平台,选中想要复制的部分后点击一下即可复制成功。路径跳转:cd 路径,进行路径跳转。查看文件内容:cat 文件名,以文本形式查看文件内容。具体见本文3.2.1.1.的使用。过滤器:要查看的文件名或目录名 | grep 要过滤的内容,过滤文件内容或者过滤目录内容。过滤文件内容可见本文3.2.1.1.的使用。修改文件内容:vim 文件名,以文本形式修改文件内容。具体见本文3.2.1.1.的使用。使用以前输入的命令:方向上下键可以使用以前输入的命令。在当前目录下打开命令行:对win有用,在文件目录名那里点击后输入cmd即可在当前目录下打开命令行窗口查看当前目录下内容:ls清空命令行窗口:clear(以前输入的命令会保留)创建目录:mkdir 目录名快速前进或后退命令的一个单词:ctrl加方向左键或右键
0.配置环境
该步骤仅是在第一次使用超算平台,或使用新的超算平台时,下载配置相关环境所用。
对于组内的学弟学妹们,本人已经配置好相关环境,可以直接从
1.激活环境开始。
# 首先运行module avail min查看具体可以安装的miniforge环境
# 有可能是miniforge3/24.11
module load miniforge/24.1.2
conda create -n demo python=3.7
# 该过程会询问是否更新一些库,全部选择更新:按“y”并回车
source activate demo
conda install numpy
conda install networkx=2.3
pip install DuIvyTools
mkdir run/你的名字缩写
# 这一步是因为该平台可能是整个组共用,如果只有单人使用,可以忽略。
1.激活环境
# 具体看安装时安装了哪个。有可能是miniforge3/24.11
module load miniforge/24.1.2
# 输入后不会有任何反应
source activate demo
# 输入后命令前会有(demo)
cd run/你的名字缩写
# 这一步是因为该平台可能是整个组共用,如果只有单人使用,可以直接cd run
# 可能是gromacs/2025.1-mpi等,具体看module avail gromacs
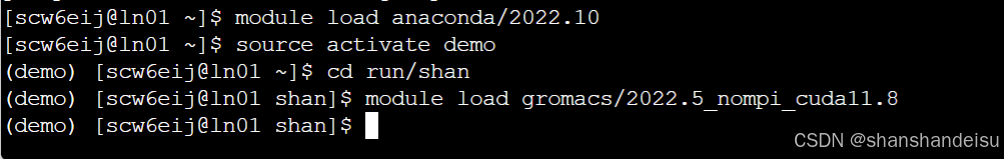
module load gromacs/2022.5_nompi_cuda11.8
# 输入后不会有任何反应
因为我本人创建的是run/shan作为工作目录,所以如下:
然后上传文件夹里所有内容,如下:
同时上传你的protein.pdb和unk.mol2。
2.准备拓扑文件
2.1.蛋白质拓扑文件准备
gmx pdb2gmx -f protein.pdb -o protein_processed.gro -ignh
选择CHARMM36力场(输入"1"或"2"),选择水模型(输入"1")
# 生成蛋白质的拓扑文件(topol.top)、结构文件(protein_processed.gro)和位置限制文件
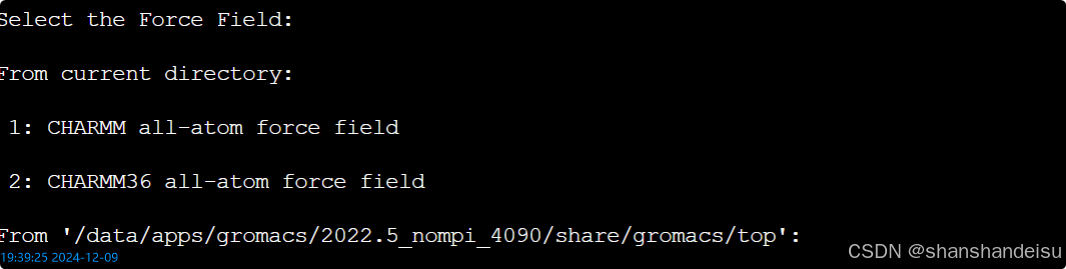
在这里可以看到我的是对应的2,显示属于我们安装的gromacs自带的。
2.2.执行脚本
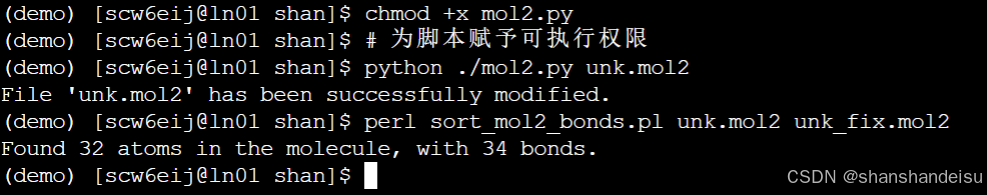
chmod +x mol2.py
# 为脚本赋予可执行权限
python ./mol2.py unk.mol2
# 执行mol2.py脚本,运行成功显示 File 'unk.mol2' has been successfully modified.
perl sort_mol2_bonds.pl unk.mol2 unk_fix.mol2
# 生成一个名为“unk_fix.mol2”的文件
综上,显示如下:
2.3.charmm-gui生成配体拓扑文件
上传小分子配体并得到相应拓扑文件。网址:https://charmm-gui.org/
2.3.1.首页
未登录时的首页如下:
记得用学术邮箱注册登录,然后首页如下:
2.3.2.提交文件
选择左侧栏的Solution Builder,拉到最下方,上传分子对接的complex.pdb文件。
报错解决:提交文件Error parsing HETAT
如果报错如下:
Error parsing HETAT, expected chain ID at column 22, but got '':

那么意味着配体的链名信息缺失。
一般来说,按照本人专栏p4的配体处理,会自动加上链名信息。
如果你不是按照该方法的话。可以选择手动加上链名信息。
或者本人在这提供额外的脚本modify_complex.bat基于参考,如下:
@echo off
setlocal enabledelayedexpansion
rem 输入和输出文件
set input_file=Structures_1.pdb
set output_file=Structures_1_copy.pdb
rem 如果输出文件已存在,删除它
if exist "%output_file%" del "%output_file%"
rem 遍历输入文件的每一行
for /f "delims=" %%A in ('type "%input_file%"') do (
set "line=%%A"
rem 检查是否以 HETATM 开头
if "!line:~0,6!" == "HETATM" (
rem 替换 UNK 后第二个空格为 D
set "line=!line: UNK = UNK D !"
)
echo !line! >> "%output_file%"
)
echo 修改完成,结果已保存到 "%output_file%"
需要根据个人情况修改input_file、output_file、链名信息。
2.3.3.选择模型/链
然后可以看到网站自动分离了蛋白和配体(Type为Protein和Type为Hetero)。
我们需要将所有都勾选上,然后页面右下角选择next step

2.3.4.操作PDB
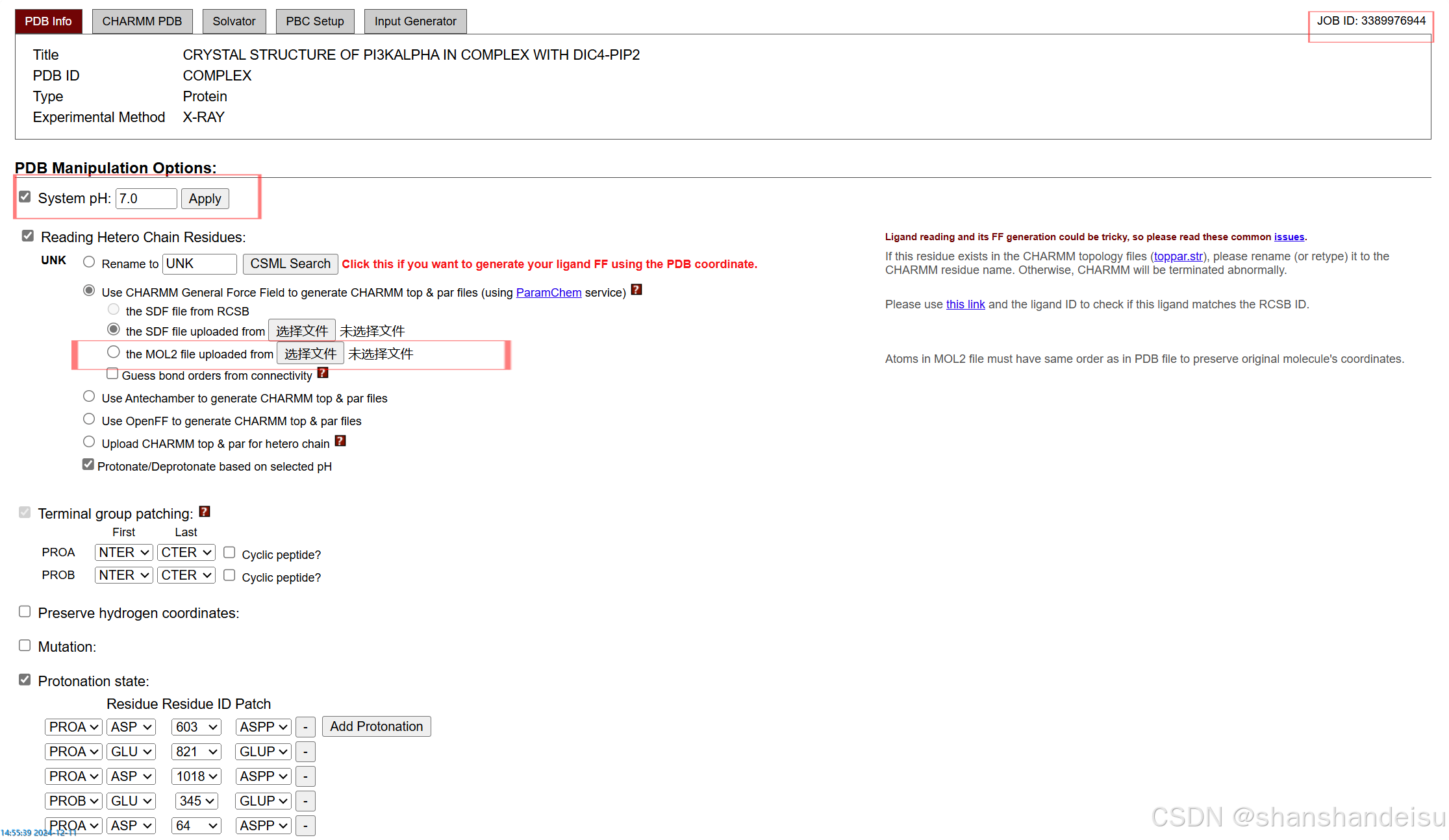
然后如果我们需要调节模拟的体系pH值,可以输入并点击Apply(我一般是调为7.35)。
并勾选上传MOL2格式的配体文件。选择Next Step。
2.3.5.生成PDB
该步骤保持默认参数即可,选择Next Step。
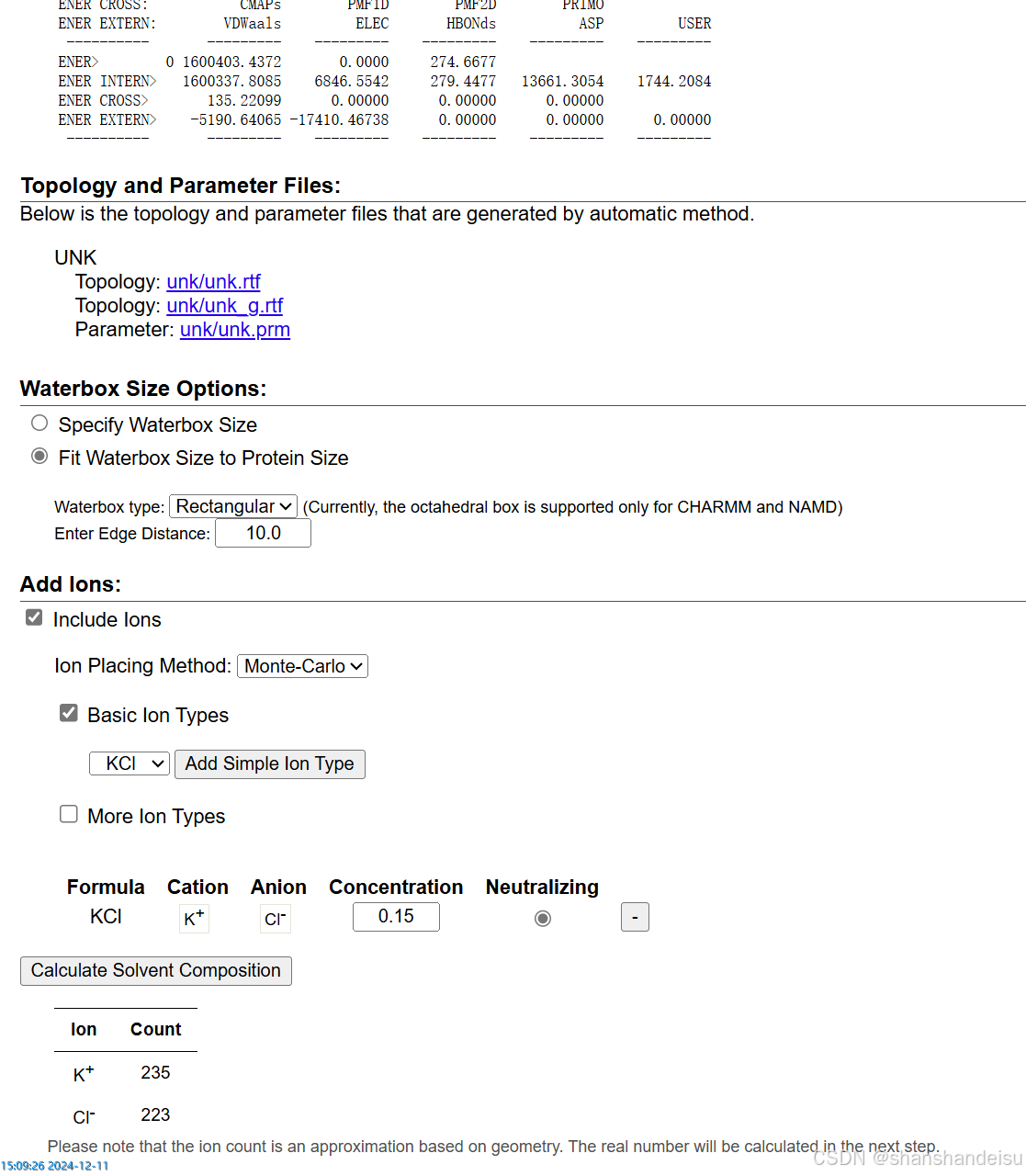
2.3.6.溶剂化分子
该步骤保持默认参数即可,选择Next Step。

这一步骤会比较久,如果没完成的话,网页页面会一直是空白的。不过我们可以关闭网页,任务会在后台服务器继续计算。
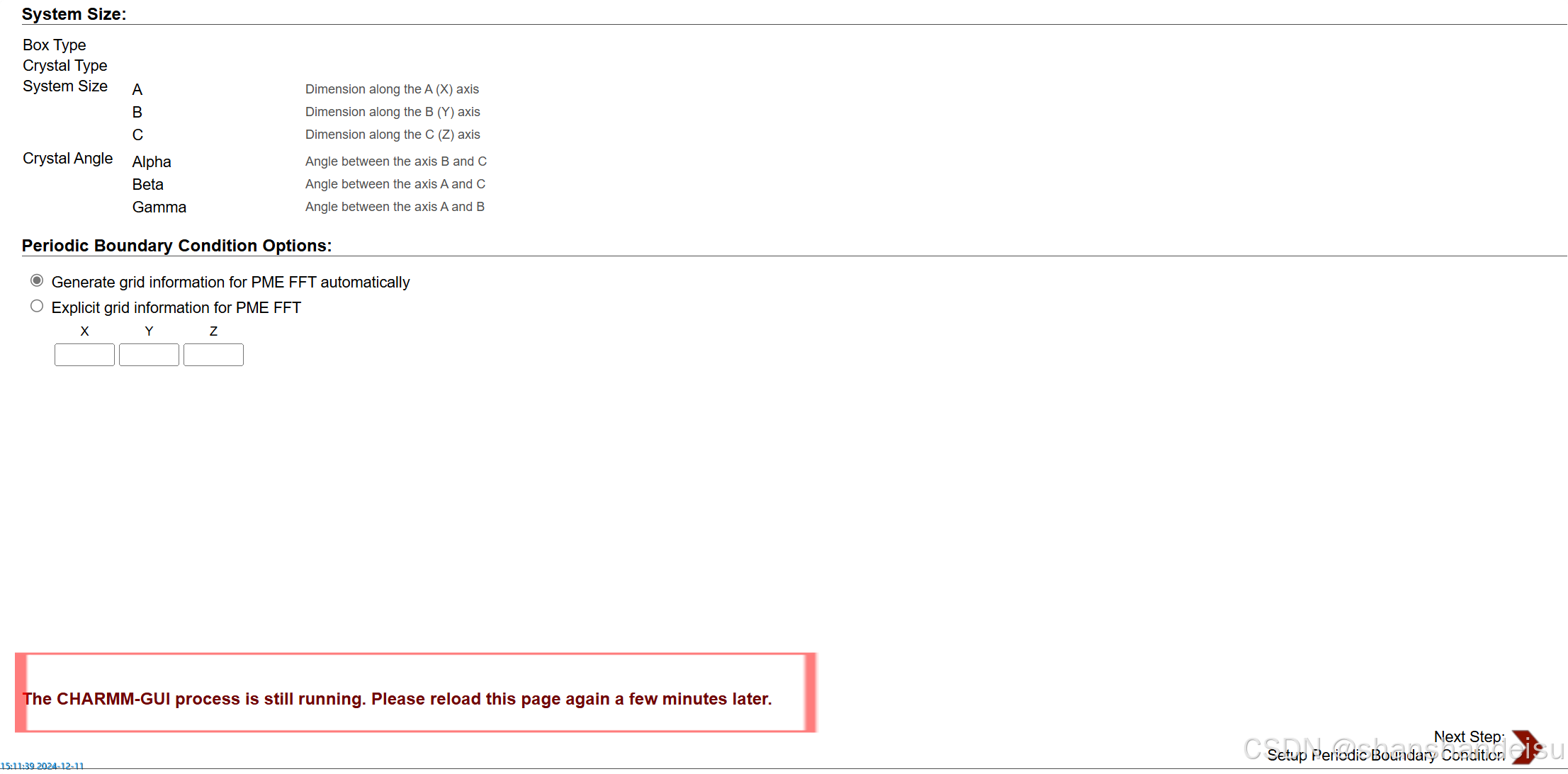
2.3.7.设置周期性边界条件
勾选GROMACS后,选择Next Step。
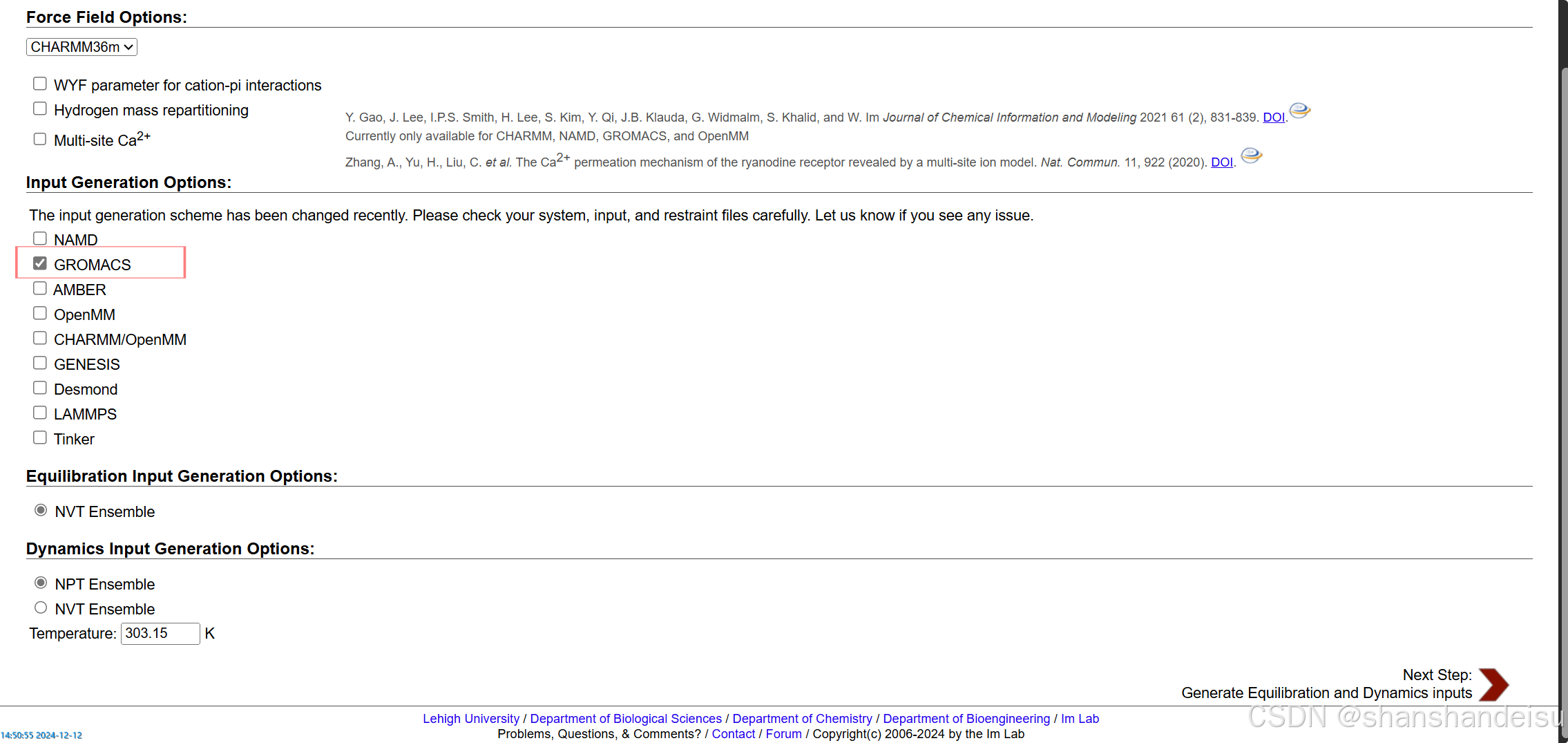
2.3.8.生成平衡和动力学输入参数
到这一步就是处理完毕了,我们点击右上角的下载即可。
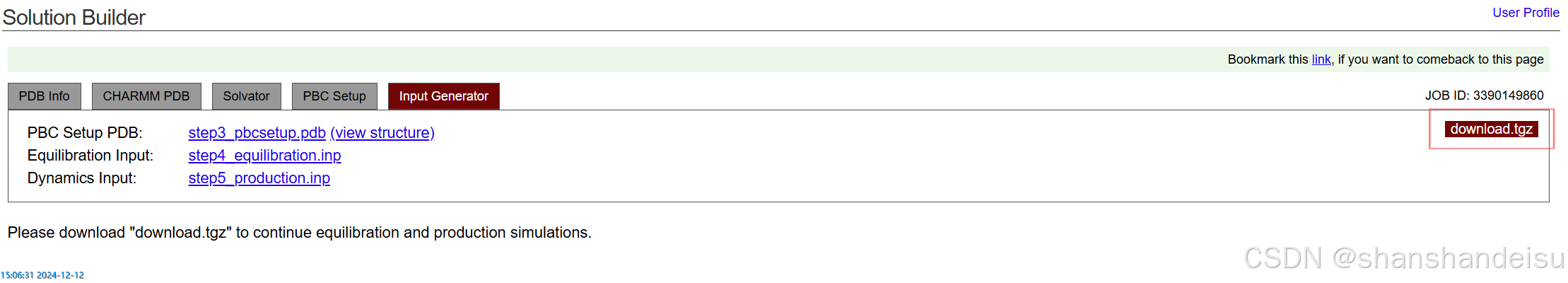
最后下载下来名为charmm-gui.tgz的压缩包,解压后会是一个charmm-gui JOB ID的文件夹,进入该文件夹后,我们找到里面子gromacs文件夹的以下文件:
(1)step3_input.gro,注意改名为:solv_ions.gro
(2)topol.top
(3)toppar文件夹
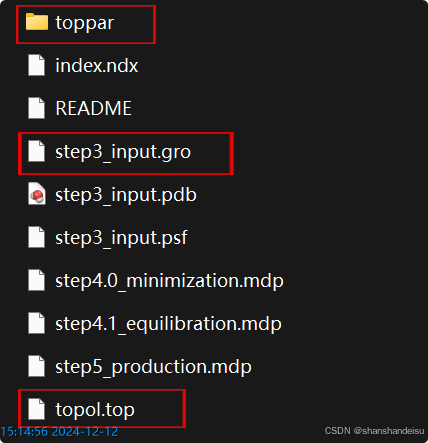
将这三者上传到超算平台上即可。
回顾任务
需要注意的是,以上每个步骤的页面右上角都存在着当前任务的JOB ID。
我们需要记住这个ID,以免某个步骤出错了,需要返工。或者某个步骤计算较久,我们将网页关闭的情况。
在任何一个步骤我们都可以关闭网页,打开job retriever,输入ID并提交。
官方只会保存任务状态一周。

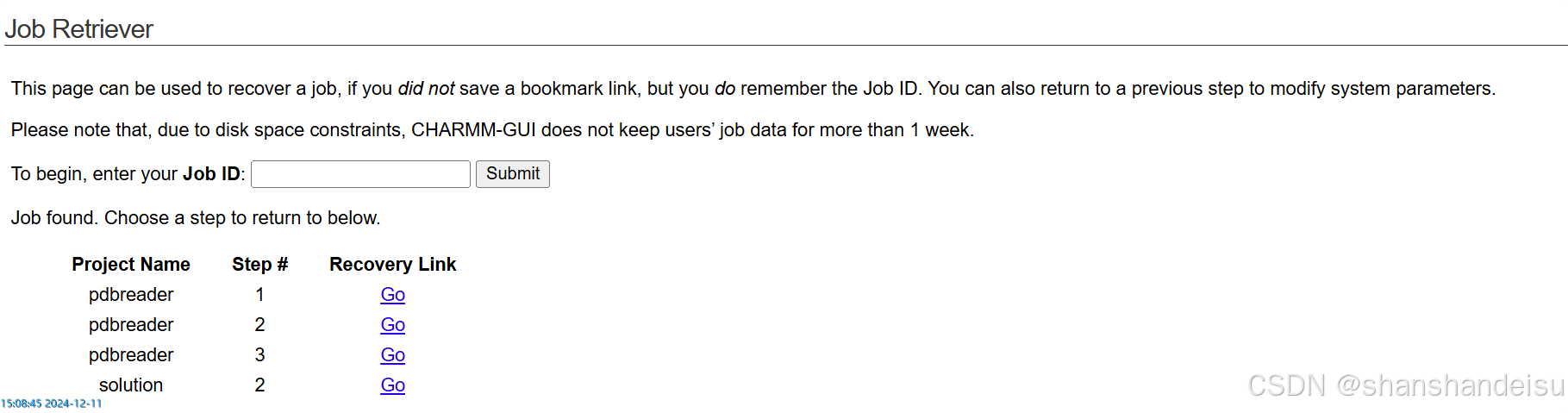
可以看到我们进行到的步骤。在这我们点击任意步骤,如果任务还在进行,就可以在页面最下方看到提示:任务还在进行。请过几分钟后重新刷新该页面。
3.动力学模拟
3.1.能量最小化
gmx grompp -f em.mdp -c solv_ions.gro -p topol.top -o em.tpr -maxwarn 2
# 给能量极小化步骤提供准备文件
gmx mdrun -v -deffnm em
# 正式能量最小化,一般2分钟左右,如果想加速的话就用以下命令,默认是8核,可以自行更改。
# gmx mdrun -v -deffnm em -nt 8
3.2.控温和控压平衡
3.2.1.建立索引文件(视情况而定)
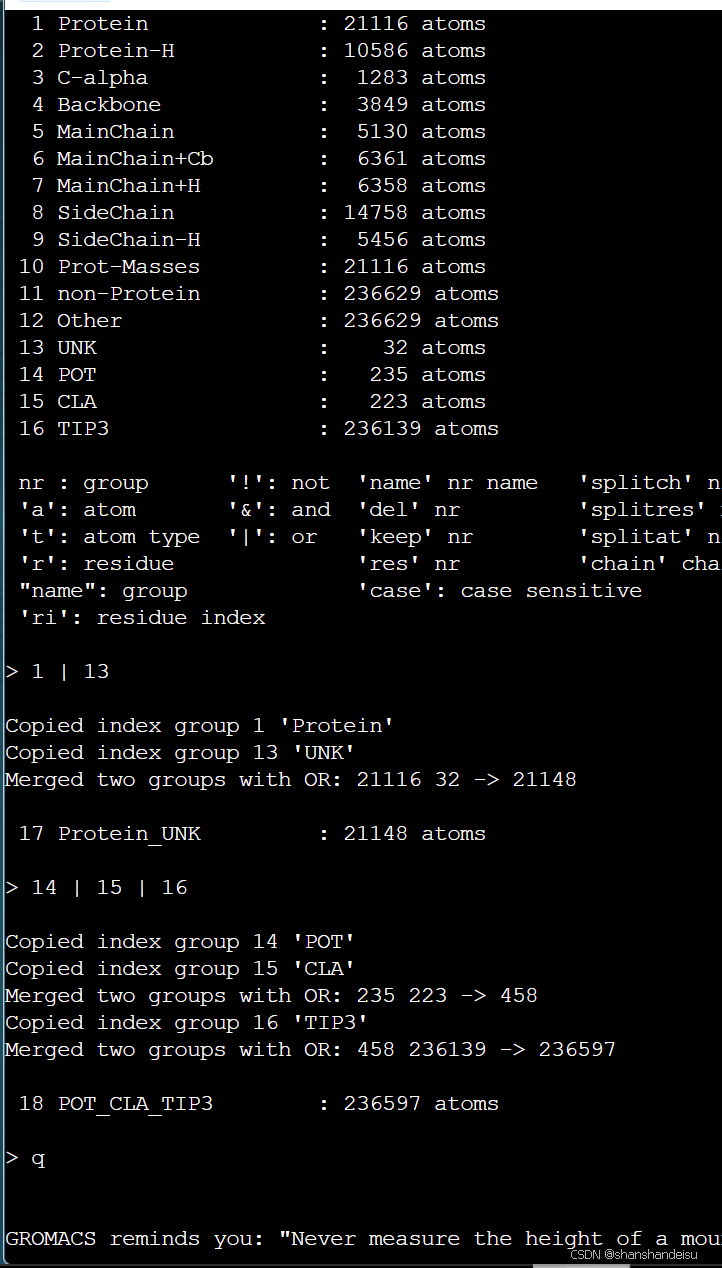
gmx make_ndx -f em.gro -o index.ndx
输入`1 | 13`后回车(对应`Protein`和`UNK`)
输入`14 | 15 | 16`后回车(对应`POT`和`CLA`和`TIP3`)
输入`q`后回车。
一般1 | 13(对应Protein和UNK)是不会变的。
在charmm-gui搭建的体系中,水分子的索引名是TIP3,氯离子是CLA,钾离子是POT。一般也不会变,因为默认加钾离子和氯离子去平衡体系的。
3.2.1.1.蛋白对接口袋含有其他离子(linux修改文件、查看文件)
但如果你的蛋白对接口袋中含有其他离子,譬如铁离子,就需要额外添加索引。
譬如它列出来的列表为:
13 UNK
14 POT
15 CLA
16 FEPEP(忘了是FE还是FEPEP了)
17 TIP3
那么就需要输入的是14 | 15 | 16 | 17
此外,还需要注意一下输入索引后它生成的索引组的顺序。
譬如在这我生成的索引组就是
POT_CLA_TIP3
但如果你的蛋白对接口袋中含有其他离子,譬如铁离子,那么生成的索引组可能如下:
POT_CLA_FEPEP_TIP3
此时我们就需要更改nvt.mdp、npt.mdp、md.mdp中的温度藕合组内容。
可以选择在自己电脑上改好了再上传到超算平台。
也可以直接在超算平台上用以下命令修改:
vim md.mdp- 输入
i后找到需要修改的地方修改 - 修改完成点击
esc退出修改, - 输入
:wq并回车,保存修改。
重复以上步骤,修改剩余mdp文件
修改完成后,输入以下命令可以查看文件内容
cat md.mdp
输入以下命令就是在过滤文件相关内容
cat md.mdp | grep tc-grps
用来确保修改正确。

可以看到,我们已经修改好了文件内容。
3.2.2.NVT平衡
gmx grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -n index.ndx -o nvt.tpr
sbatch -p gpu_4090 --gpus=1 nvt.sh
# 使用slurm脚本将作业提交至计算节点运行,默认使用1个gpu,嫌弃速度慢可以改为2个。注意和组内其他同学沟通下
squeue
# 执行squeue命令查看任务运行状态,如果不显示任务条目,则说明任务运行完毕
3.2.3.NPT平衡
gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -r nvt.gro -p topol.top -n index.ndx -o npt.tpr
sbatch -p gpu_4090 --gpus=1 npt.sh
squeue
3.3.正式模拟
如果你不需要修改模拟的条件,直接运行以下命令(默认是模拟100ns):
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -n index.ndx -o md.tpr
sbatch -p gpu_4090 --gpus=8 md.sh
# 这一步因为耗时最久,所以我一般8个gpu拉满。注意和组内其他同学沟通下gpu使用情况。
但如果你需要修改模拟时间,那么按照3.2.1.1.的方法修改md.mdp中的nsteps即可

4.计算结果
4.1.矫正轨迹
gmx trjconv -s md.tpr -f md.xtc -o md_nojump.xtc -center -pbc nojump -ur compact
# 提示选择什么作为视觉中心,选择1,Protein。提示选择什么作为输出体系,选择0,Sytem
选择Protein后回车,再选择System后回车
gmx trjconv -s md.tpr -f md_nojump.xtc -o md_noPBC.xtc -center -pbc mol -ur compact
选择Protein后回车,再选择System后回车
gmx trjconv -s md.tpr -f md_noPBC.xtc -o md_fit.xtc -fit rot+trans
选择Backbone后回车,再选择System后回车
4.2.RMSF计算
gmx rmsf -s md.tpr -f md_noPBC.xtc -n index.ndx -o fws-rmsf.xvg -ox fws-avg.pdb -res -oq fws-bfac.pdb
选Protein后回车
RMSF出错解决方案
为什么需要先计算RMSF呢?
因为你的蛋白是单聚体的话,基本都不会出错。
但如果蛋白是多聚体(也就是多链)的话,可能RMSD、氢键、自由能形貌图等都是正确的,但是因为RMSF的横坐标是残基序号,所以导致错乱。
因此建议先计算RMSF,得到fws-rmsf.xvg后去qtgrace进行可视化,检查一下。
如果RMSF出错的话,解决方案如下:
- 修复蛋白结构。
- 尝试其他命令去做
4.1.矫正轨迹。具体见该博客:https://www.jianshu.com/p/5dc493663ed2。 - 固定结合区域的周边氨基酸位置,避免跑飞。
4.3.RMSD计算
# 计算蛋白骨架,时间为ns的RMSD
gmx rms -s md.tpr -f md_noPBC.xtc -o rmsd.xvg -tu ns
选择4后回车,再选择4后回车
# 计算蛋白骨架,时间为ps的RMSD
gmx rms -s md.tpr -f md_noPBC.xtc -o rmsd_ps.xvg
选择4后回车,再选择4后回车
# 计算配体的RMSD
gmx rms -s md.tpr -f md_noPBC.xtc -n index.ndx -o rmsd_Lig.xvg -tu ns
选择13后回车,再选择13后回车。
4.4.回旋半径gyrate计算
gmx gyrate -s md.tpr -f md_noPBC.xtc -o gyrate.xvg
选择4后回车
4.5.氢键计算
gmx hbond -f md_noPBC.xtc -s md.tpr -num hbond_num.xvg
选择Protein后回车,选择UNK后回车
# 这一步会需要比较久时间,约5分钟,可以选择以下代码替代(使用8核并行计算加速)
gmx hbond -f md_noPBC.xtc -s md.tpr -num hbond_num.xvg -nt 8
4.6.自由能形貌图
chmod +x pc_combine.py
python ./pc_combine.py rmsd_ps.xvg gyrate.xvg output.xvg
# 输出done
gmx sham -tsham 310 -nlevels 100 -f output.xvg -ls gibbs.xpm -g gibbs.log
dito xpm_show -f gibbs.xpm -3d -ip -o gibbs_3d.pdf
dito xpm_show -f gibbs.xpm -ip -o gibbs.pdf
4.7轨迹动图和MMPBSA(可选)
# 生成轨迹动图
gmx trjconv -s md.tpr -f md_fit.xtc -n index.ndx -o movie.pdb -dt 100
选索引组后回车
# 生成MMPBSA
source activate gmxMMPBSA
sbatch -N 10 -p c-16-1 -n 160 -c 1 gmx_MMPBSA.sh
5.结果可视化
对于RMSD、RMSF之类的都是扔到qtgrace里面,在这不过多赘述。
对于自由能形貌图,我们4.计算结果得到的是2D和3D的PDF格式。
红色代表能量高的构象,蓝色代表能量低的构象。能量越低越稳定。
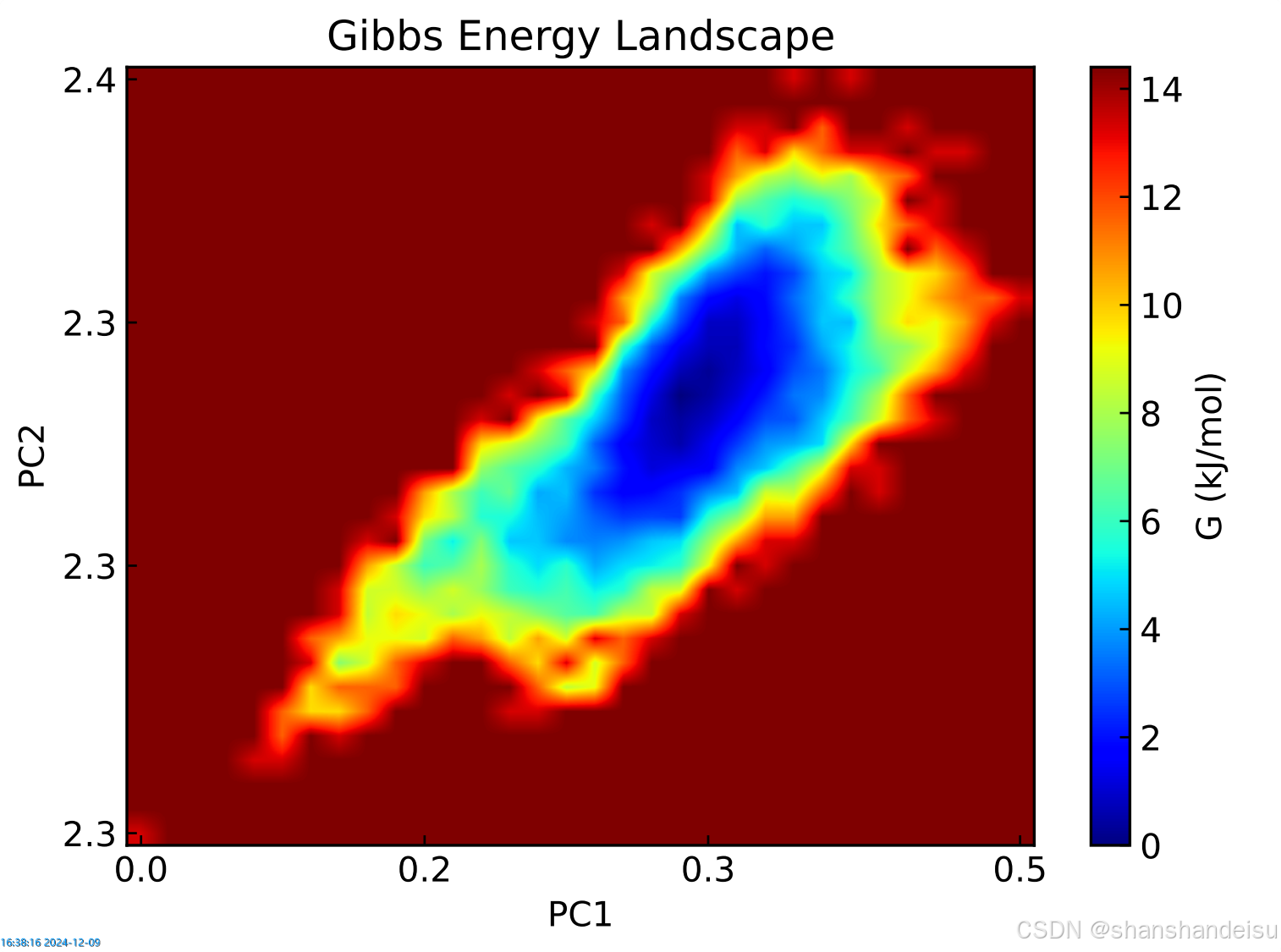
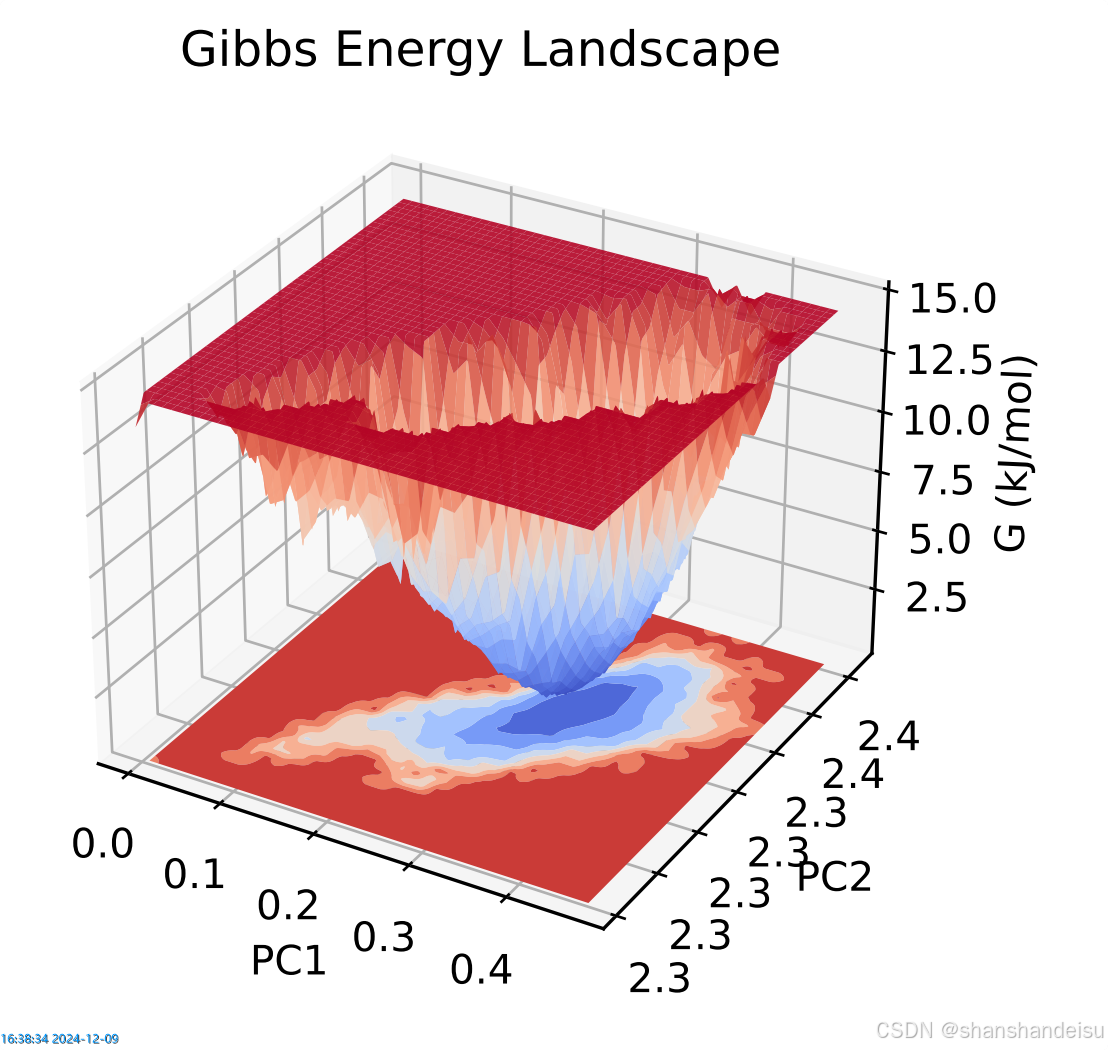
对于轨迹动画movie.pdb,直接pymol,然后在命令行里面输入如下:
intra_fit movie
smooth
smooth
然后preset->ligand site/cartoon即可,一般需要3-5分钟。
本文地址:https://www.vps345.com/5340.html